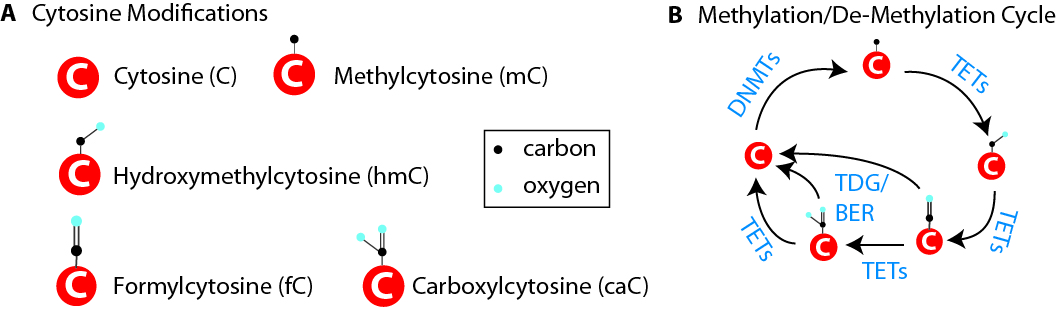
As research extends into the role of neuroepigenetics in normal and pathological conditions researchers are developing new techniques and data analysis methods for analysis of DNA modifications, namely, cytosine methylation and hydroxymethylation. While older techniques for modification analysis performed relative quantitation across regions of the genome or examined average genome levels, these analyses lack the desired specificity and rigor. With technological advances cytosine modifications can now be readily analyzed in an absolutely quantitative, base-specific manner at both CpG and non-CpG sites throughout the genome by next generation sequencing. Recently, we and others have developed techniques that allow analyses to be focused to specific regions of the genome for directed hypothesis testing in aging research. We continue to develop sequencing methods and bioinformatics tools for generating these large data sets and converting them into biological insights.